Unlike restriction enzymes recognizing palindrome sequences, type IIS restriction enzymes recognize asymmetric sequences and cut outside the recognition sequences. Golden Gate Assembly capitalizes on variable combinations of sticky ends made by type IIS restriction enzymes and allows more than 30 fragments to be ligated together in one reaction. However, we rarely need to combine that many fragments, and we usually go with Gibson assembly for joining a small number of fragments.
On the other hand, Golden Gate Assembly is perfect for introducing variable small fragments of DNA, such as guide RNAs, into a common vector backbone. It is convenient to parallel process multiple guide RNAs by ordering pairs of DNA oligos with matching sticky ends to vectors linearized by type IIS enzymes. Here, we demonstrate an example of introducing Cas9 guide RNAs to the lentiCRISPR v2 vector.
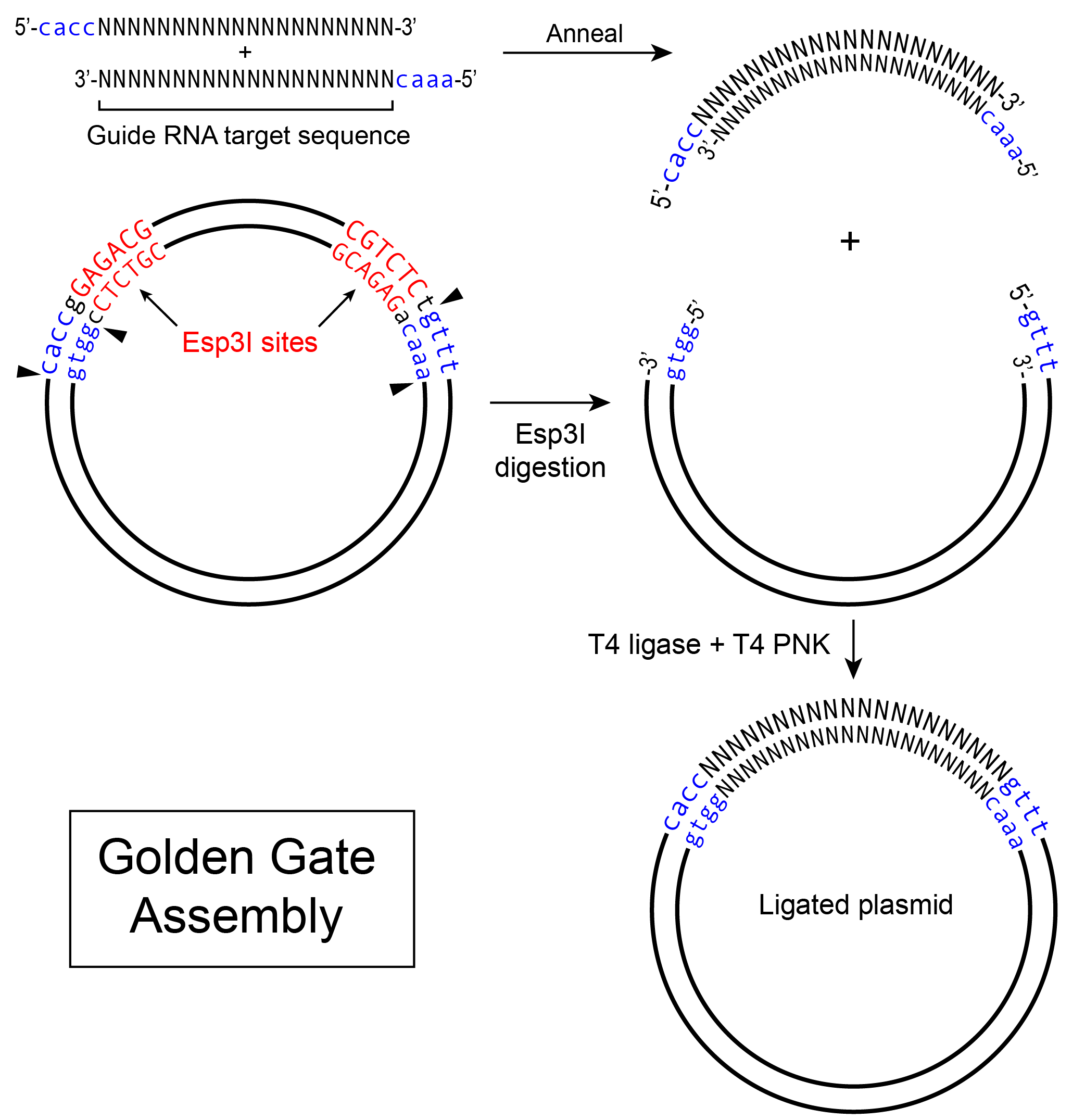
1. Linearizing vectors
-
Linearize the lentiCRISPR v2 vector with Esp3I (isoschizomer of BsmBI). Set up a 50 μL digestion reaction in a 1.5 mL tube:
Stock for 50 µL Water 34.5 10× CutSmart Buffer 5 Plasmid (1 µg/µL) 10 Esp3I (10 U/µL) 0.5 -
Incubate the reaction at 37°C for 1-2 hours.
-
During the incubation, make 1% agarose gel with 0.0001% crystal violet (stock is 0.2%, use at 2000x) for gel extraction.
-
After incubation, run the reaction using the crystal violet gel at 100V for 20-30 min.
- For the lentiCRISPR v2 vector, we are expecting 2 bands at 13 kb and 1873 bp (filler sequence).
-
Cut the larger band at 13 kb and purify with the Qiagen gel purification kit.
- Purified DNA fragments can be stored at -20°C and used for multiple freeze-thaws.
2. Annealing oligo pairs
-
For a small number (<5) of oligo pairs, anneal them in individual 1.5 mL tubes. For a larger number of oligo pairs, anneal them in a 96-well PCR plate.
- In the latter case, order oligo pairs in formats of 100 μM solutions in a 96-well plate and use a multichannel pipette to transfer and mix oligos.
-
For each oligo pair, mix 10 μL oligo 1 + 10 μL oligo 2 + 80 μL water (10 μM duplex). Heat the tubes or 96-well PCR plate at 95°C for 5 mins. Then cool down on bench for 15-30 mins to form oligo duplexes.
-
In a new set of tubes or wells in the 96-well PCR plate, add 200 µL water in each tube or well.
-
Transfer 1 µL of each 10 μM duplex to the new tubes or wells to obtain a final concentration of 0.05 μM.
3. Ligation
-
We use a combination of the T4 ligase and T4 PNK for this type of ligation.
-
Make a master mix (N+1 or +2) without the oligo duplex, and aliquot 19 μL into a new set of tubes or wells. Each 20 μL reaction contains:
Stock for 20 µL ×14 Water 13 182 Digested vector (50 ng/µL) 1 14 10× T4 ligase buffer 2 28 T4 PNK 2 28 T4 ligase 1 14 0.05 μM oligo duplex to add to add -
Transfer 1 μL of each diluted oligo duplex into these tubes or wells.
- Use a multichannel for a large number of reactions in the 96-well plate format.
-
Ligate at room temperature for 10-15 min.
- Thaw competent cells when starting the ligation reaction.
- Warm up sufficient ampicillin selection plates at 37°C.
4. Transformation
It is usually sufficient to transform 1 µL ligation reaction using 10-20 µL commercially available chemically competent cells. For lentiviral vectors, we use chemically competent cells such as NEB Stable Competent E. coli cells:
- Turn on a water bath set at 42°C.
- Thaw a sufficient number of competent cell aliquots (50 µL/tube) on ice for 10-15 min. 1. Aliquot 10-20 µL competent cells in a new set of tubes or wells.
- Mix 1 µL reaction into each aliquot of competent cells.
- Incubate on ice for 10 min. Do not mix.
- Heat shock at 42°C for exactly 30 sec.
- Place on ice for 3 min.
- Add 50 µL clean broth (e.g., LB) to each tube or well, and spread cells onto an ampicillin selection plate.
- Grow at 37°C overnight or 30°C for 24 h (for plasmids with high risk of recombination).