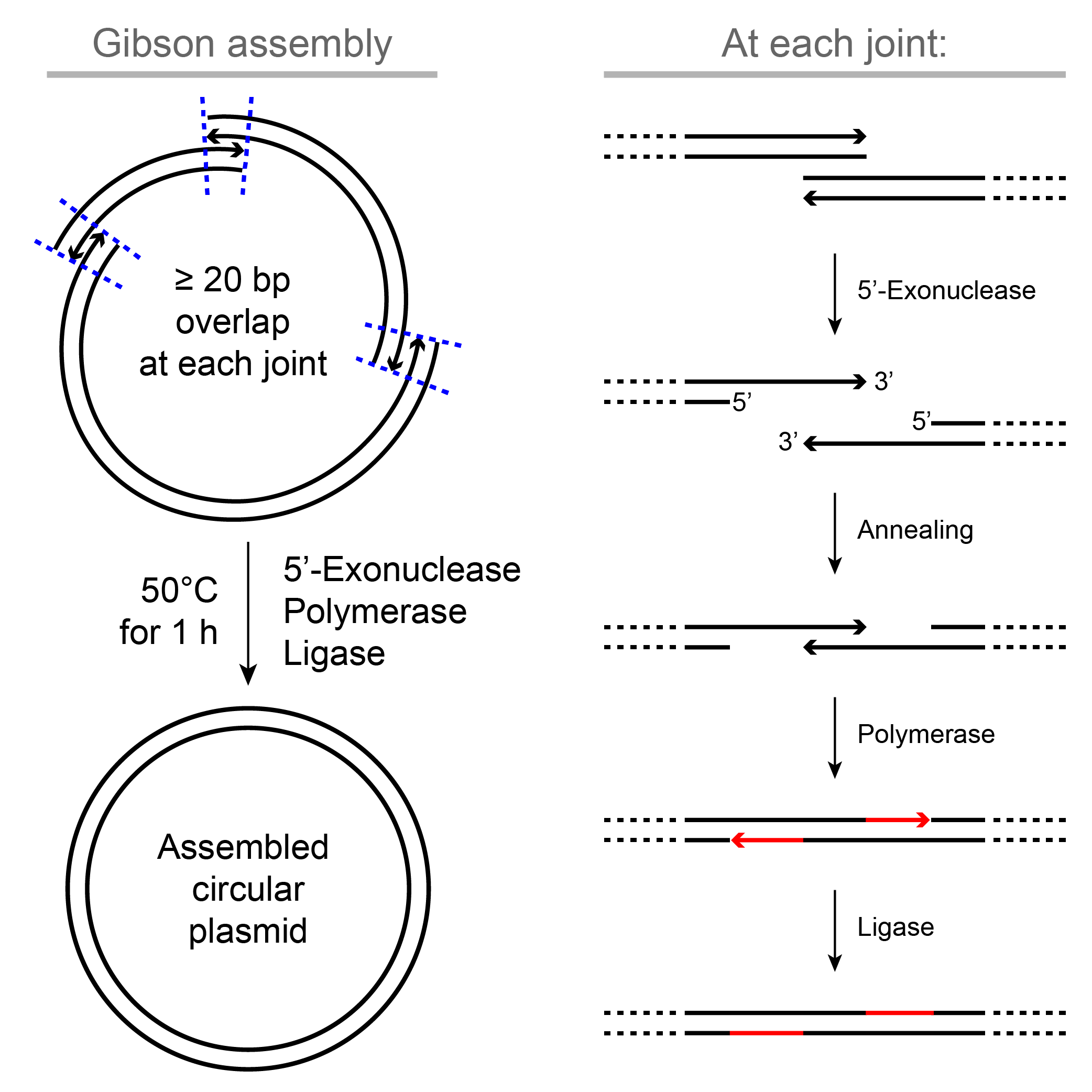
Gibson Assembly uses a mixture of DNA 5’-exonuclease, polymerase and ligase to ligate 2~6 fragments with ≥ 20 bp overlapping ends into a circular plasmid in one step of 1-hour incubation at 50°C. This method gives us a great deal of freedom and allows scarless ligation of arbitrary fragments, which can be obtained from PCR, enzymatic digestion or synthesis.
1. Prepare the Gibson Assembly Mix
We use homemade Gibson Assembly mix, but you can also buy NEB Gibson Assembly Mix. The following recipe is modified from OpenWetWare and Rabe and Cepko 2020.
-
Prepare 5× Isothermal Reaction Buffer (IRB). Divide into 500 uL aliquots and store at -20°C.
Stock for 3 mL for 6 mL PEG 8000 750 mg 1.5 g 1 M Tris-HCl, pH 7.5 1.5 mL 3 mL 1 M MgCl2 150 µL 300 µL 1 M DTT 150 µL 300 µL 100 mM each dNTP 4× 30 µL 4× 60 µL 100 mM NAD 150 µL 300 µL Water ~250 µL ~500 µL -
Make the 1.33× Assembly Master Mix (AMM). Divide into 60 µL aliquots and store at -20°C.
- AMM can be frozen and thawed for >10 times without obvious drop in activity.
- ET SSB was shown to greatly enhance the efficiency by Rabe and Cepko 2020. Classical recipe does not have ET SSB.
- Caution: DO NOT replace Phusion with Q5 DNA polymerase. In one such attempt, the AMM yielded no colonies for a few straightforward assembly reactions. Going back to Phusion worked as expected.
Stock for 300 µL for 600 µL Water 170.9 µL 341.8 µL 5× IRB (see above) 80 µL 160 µL Taq DNA ligase (40 U/µL) 40 µL 80 µL Phusion DNA polymerase (2 U/µL) 5 µL 10 µL T5 exonuclease* (1 U/µL) 1.6 µL 3.2 µL ET SSB (500 µg/ml) 2.5 µL 5 µL *T5 exonuclease was diluted to 1 U/µL in storage buffer (50 mM Tris-HCl, 100 mM NaCl, 1 mM DTT, 0.1 mM EDTA, 50% Glycerol, 0.1% Triton X-100, pH 7.5 @ 25°C) and stored at -20°C. To make storage buffer:
Stock for 1000 µL Water 423.8 µL Glycerol 500 µL 1 M Tris-HCl, pH 7.5 50 µL 5 M NaCl 20 µL 1 M DTT 1 µL 0.5 M EDTA 0.2 µL 20% Triton X-100 5 µL
2. Design and obtain DNA fragments
We highly recommend the free software ApE for everyday editing of DNA sequences.
-
Design your desired construct in ApE by piecing together different fragments.
-
Identify up to 6 fragments that you can obtain by PCR amplification, enzymatic digestion from an existing plasmid or by direct synthesis (up to 3 kb; avoid complex features such as repetitive elements).
- Using enzymatic digestion can avoid introducing mutations during PCR amplification, but it could be hard to get enough for small fragments. It should be used when very large fragments need to be used (e.g. > 10 kb).
- PCR amplification should use high fidelity DNA polymerase such as the NEB Q5 to minimize mutations.
- For DNA fragment synthesis, we use the IDT gBlocks gene fragments service.
-
Design primers or synthesized DNA sequences to have at least 20 bp overlapping sequences at each joint.
-
For PCR amplification with Q5:
Stock for 50 µL 2× Q5 master mix 25 µL Water 18 µL Primer 1 2.5 µL Primer 2 2.5 µL Template (10-20 ng/µL) 2 µL -
DNA fragments by PCR amplification and enzymatic digestion should be gel purified following this protocol.
3. Gibson assembly of DNA fragments
-
In a PCR tube, make 2.5 µL of an equi-molar mix of all DNA fragments. If a fragment is <300 bp, double or triple its amount. Use this Google Spreadsheet to calculate volumes to mix by entering fragment lengths and concentrations of purified fragments.
- To avoid pipetting tiny volumes, make a 5 µL mix and aliquot 2.5 µL for the reaction. If the DNA concentration is too high (>100 ng/µL), dilute the stock first.
- According to OpenWetWare, 5-50 ng of each ~6 kb DNA should be used per 10 µL reaction.
- I have used anywhere between 25-150 ng of each ~6 kb DNA per 10 µL reaction. Within this range, more DNA does not seem to adversely affect the reaction.
-
Add 7.5 µL of 1.33× Assembly Master Mix for a 10 µL reaction. Incubate at 50°C for 15-60 min.
- 1 hour incubation is generally regarded as optimal. For 2-fragment assembly, 30 min works, and 15 min may be OK as well.
4. Transformation of Gibson assembly reaction
Gibson assembly reactions are salty, so for transformation by electroporation with electrocompetent cells, it need to (1) using a very small volume (<1 µL reaction per 25 µL competent cells), (2) diluted with water, or (3) pelleted with ethanol and resuspended in water. On the other hand, transformation with chemically competent cells can be done as usual.
For a typical transformation with chemically competent cells such as NEB Stable Competent E. coli cells:
- Turn on a water bath set at 42°C.
- Thaw a tube of 50 µL chemically competent cells on ice for 10 min.
- Mix 5 µL Gibson assembly reaction with 50 µL competent cells.
- Incubate on ice for 10 min. Do not mix.
- Heat shock at 42°C for exactly 30 sec.
- Place on ice for 3 min.
- Spread onto a selection plate. Grow at 37°C overnight or 30°C for 24 h (for plasmids with high risk of recombination).
For exceptionally large plasmids (> 20 kb), electroporation may work better due to higher efficiency and higher chance to take in the large complete assembled plasmid DNA in the reaction.
For a typical electroporation with electrocompetent cells such as Endura™ ElectroCompetent Cells:
- Pre-chill one sterile 1.5 mL tube and 1.0 mm cuvettes for each transformation on ice. There is no need to label them.
- Thaw electrocompetent cells completely on ice (10-20 min).
- Make sure there is a saved setting on the electroporator: 10 µF, 600 Ohms and 1800 Volts. Make it ready to go.
- Pre-warm sufficient recovery medium (1 mL per reaction) and selection plates at least to room temperature.
- Pre-label room-temperature sterile 1.5 mL tubes for each transformation for recovery. Get 1 mL pipet and sterile tips ready.
- On ice, aliquot 25 µL competent cells into each 1.5 mL tube.
- On ice, pipet 0.5 µL Gibson assembly reaction into one aliquot of competent cells. Do not pipet to mix. Immediately transfer the entire 25 µL mixture into a cuvette.
- Use a KimWipe to wipe dry the outside of the cuvette, place it into the electroporation chamber and start the electroporation.
- Once the electroporation is started, pipet up 1 mL recovery medium.
- Immediately after the electroporation is done, add in 1 mL recovery medium and transfer into pre-labeled 1.5 mL tubes.
- Recover in a shaking incubator at 250 rpm for 1 hour at 37°C by taping the tube horizontally on the bottom of the shaker.
- Spread 100 µL on a selection plate. Keep the remaining 900 µL at 4°C.
Note:
- You can of course also use 10 mL Falcon tubes for recovery, but 1.5 mL tubes work just fine.
- If an arc is observed during electroporation, repeat the transformation with a new aliquot of cells by using water-diluted Gibson assembly reaction to reduce salt concentration. Alternatively, also dilute the competent cells 1:1 with cold sterile water to further reduce the overall salt concentration.
5. Troubleshooting and tips
- No or very few colonies
- Double check your design: do all joints have ≥ 20 bp overlapping ends?
- Use higher concentration DNA in the reaction, if some fragments were < 10 ng/µL.
- Use competent cells with higher transformation efficiency.
- For very large constructs (> 20 kb), use electroporation instead of chemically competent cells.
- It is common to have only a few colonies for very large constructs (> 20 kb), and they often turn out to be correct.
- High false-positive or mutation rate
- Use gel purification for PCR-amplified fragments.
- Re-design ends of DNA fragments, if different joints have highly similar sequences.
- When using synthesized DNA, use it directly instead of PCR amplification from it.
- What if I need to assemble an array of cassettes with identical ends (e.g., multiple guide RNA expressing cassettes)?
- Use unique 20-bp adaptor sequences to join the different cassettes. I often use a few lacZ-derived adaptors.
- What if I need to assemble more than 6 fragments (e.g., 10 or 18 fragments)?
- Do it stepwise with intermediate constructs. First assemble ≤ 6 fragments per construct into intermediate constructs, and then PCR amplify or cut out assembled parts to further assemble into the desired construct.